ژنتیک کاردیومیوپاتی هیپرتروفیک: نکات کلیدی
نکات کلیدی زیر از مقالهای درباره ژنتیک کاردیومیوپاتی هیپرتروفیک (HCM) که به بررسی پیامدهای تثبیتشده و نوظهور برای عملکرد بالینی میپردازد، قابل یادآوری است:
- شایعترین ژنهای شناساییشده در HCM سارکومری غیرسندرمی شامل MYH7 (کدکننده زنجیره سنگین بتا-میوزین)، MYBPC3 (پروتئین C متصلشونده به میوزین)، TNNT2 (تروپونین T) و TNNI3 (تروپونین I) هستند. این ژنها ۹۰% موارد HCM با ژنوتیپ مثبت را تشکیل میدهند.
- یافتن یک جهش بیماریزا یا احتمالاً بیماریزا (P/LP) در ژنهای شناختهشده برای ایجاد HCM، قطعیت تشخیصی را بهبود میبخشد. بر این اساس، تمام دستورالعملها آزمایش ژنتیک را برای HCM توصیه میکنند.
- آزمایش ژنتیک آبشاری در خویشاوندان بیماران مبتلا به HCM که دارای جهش P/LP شناساییشده هستند، میتواند به تعریف خطر ابتلا به HCM در آنها کمک کرده و غربالگری بیماری را هدایت کند. دفعات غربالگری در خویشاوندان به سن آنها بستگی دارد، از جمله آزمایش سالانه در دوران نوجوانی و اوایل بزرگسالی و هر ۳-۵ سال در مراحل بعدی بزرگسالی. شناسایی یک واریانت P/LP همچنین امکان آزمایش ژنتیک پیش از لانهگزینی با لقاح آزمایشگاهی را فراهم میکند.
- نفوذ بیماری در خویشاوندان با ژنوتیپ مثبت فرد مبتلا به HCM، با جنسیت مذکر و وجود ناهنجاریهای الکتروکاردیوگرام، بالاتر است. TNNI3 در مقایسه با MYBPC3 کمترین نفوذ را دارد. مرگ ناگهانی قلبی در افراد با ژنوتیپ مثبت بدون فنوتیپ HCM مشاهده نشده است.
- در غیاب هیپرتروفی بطن چپ (LVH)، حاملان واریانت ممکن است دچار اختلال عملکرد دیاستولیک، فیبروز، کریپتهای میوکارد، لتهای میترال کشیده، نقصهای پرفیوژن میوکارد و ناهنجاریهای الکتروفیزیولوژیک باشند. در صورت وجود این یافتهها، غربالگری بالینی دقیقتر برای توسعه LVH با فرکانس هر ۶-۱۲ ماه توصیه میشود.
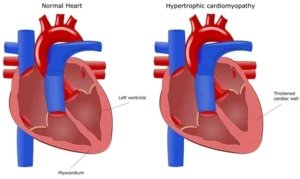
- افراد سارکومر-مثبت مبتلا به HCM در سنین پایینتری نسبت به افراد سارکومر-منفی ظاهر میشوند، هیپرتروفی شدیدتر، انسداد کمتر شایع مجرای خروجی بطن چپ، بار اسکار بیشتر و افزایش خطر آریتمی و نارسایی قلبی دارند. با این حال، ارتباطات ژنوتیپ-فنوتیپ از نظر بالینی برای گنجاندن در الگوریتمهای پیشبینی خطر چالشبرانگیز بوده است، زیرا پیشبینیکنندهها شامل عوامل بالینی مانند سن و حداکثر ضخامت دیواره هستند که حداقل تا حدی با ژنوتیپ همبستگی دارند.
- فنوکپیهای HCM شامل بیماری فابری، آمیلوئیدوز (TTR)، سندرم PRKAG2، بیماری دانون و راسوپاتیها مانند سندرم نونان هستند. این بیماریها را میتوان از سن شروع، سابقه، معاینه فیزیکی، وجود علائم خارج قلبی و نحوه وراثت متغیر برای برخی، متمایز کرد. آزمایش ژنتیک بین تمام این فنوکپیها تمایز قائل میشود که پیامدهایی برای مدیریت بالینی دارد (مثلاً تافامیدیس برای آمیلوئیدوز TTR).
- تقریباً ۶۰% بیماران HCM واریانت سارکومری قابل شناسایی ندارند. دادهها نشان میدهد که توسعه HCM در این افراد ممکن است تحت تأثیر عوامل محیطی و اثرات پلیژنیک باشد. خویشاوندان این افراد با “HCM غیرخانوادگی” ممکن است نیازی به غربالگری به دفعات افراد با ژنوتیپ مثبت نداشته باشند و ممکن است از مدیریت عوامل خطر قلبی مانند فشار خون بالا بهرهمند شوند.
- درمانهای اختصاصی بیماری اکنون برای HCM به عنوان مهارکنندههای میوزین قلبی از جمله ماواکامتن و افیکامتن در این رده در دسترس هستند. در آزمایشهای بیماران مبتلا به HCM انسدادی علامتدار، ماواکامتن و افیکامتن ظرفیت ورزشی و علائم را در مقایسه با دارونما بهبود بخشیدند. آزمایش دیگری کاهش نسبت بیماران نیازمند درمان کاهش سپتوم با ماواکامتن را نشان داد.
- آزمایشهای ژندرمانی در مراحل اولیه آزمایش برای HCM هستند و از وکتورهای مرتبط با آدنو برای DNA یا نانوذرات لیپیدی برای RNA استفاده میکنند. عوارض احتمالی ژندرمانی شامل اثرات خارج از هدف که باعث جهشزایی سلولهای سوماتیک و افزایش خطر سرطان میشود، ایمنیزایی وکتور ویروسی، تحویل ناکافی به کاردیومیوسیتها و خنثیسازی به دلیل آنتیبادیهای ناشی از عفونتهای قبلی آدنوویروس است.
https://academic.oup.com/eurheartj/advance-article/doi/10.1093/eurheartj/ehae421/7710314